
ML065 : LDR (Locus Depression) Modulator
ML065
Target Name
Locus Depression
Target Alias
LDR
Target Class
Epigenetic Enzyme
Mechanism of Action
Modulator of LDR
Biological / Disease Relevance
Epigenetics
In vitro Activity
IC50Inactive Control
Available
Target Information
Over the last several years, a few compounds have been identified that inhibit the methylation or de-acetylation pathways mediated by DNA methyltransferases and histone deacetylases. These compounds have immediate application in the treatment of cancers, as they reactivate aberrantly silenced tumor suppressor genes. There is clear need to identify additional small molecules that interact with these enzyme families, as well as new targets involved in the epigenetic control of gene expression. Our overall goal is to expand the available repertoire of small molecules that modulate gene expression and to evaluate their basic mechanism of action.
The Locus Derepression (LDR) assay detects the derepression of a GFP reporter that is stably integrated in a region of the genome of murine c127i mammary cells, which is presumably silenced. GFP transcription in this construct is controlled by a CMV promoter, which is normally strong and constitutively active. However, this line was selected for lack of constitutive expression of the GFP protein. GFP production can be induced by incubating the cells with histone deacetylase or DNA methyltransferase inhibitors. Compounds that cause derepression of the locus to express GFP are identified by enumerating GFP positive cells using a laser-scanning microplate cytometer.
Properties
ML065
NCGC00059731
| Physical & chemical properties | ||||
|---|---|---|---|---|
| Molecular Weight | 272.36534 [g/mol] | |||
| Molecular Formula | C15H16N2OS | |||
| cLogP | 2.4 | |||
| PSA | 33.2 | |||
| Storage | ||||
| Solubility | ||||
| CAS Number | ||||
SMILES:
O=C(N1CCCC1)CSC2=CC=CC3=CC=CN=C23
InChI:
InChI=1S/C15H16N2OS/c18-14(17-9-1-2-10-17)11-19-13-7-3-5-12-6-4-8-16-15(12)13/h3-8H,1-2,9-11H2
InChIKey:
RQGNWRSISVFFMW-UHFFFAOYSA-N
Activity
Summary activity statement /
ML064 (SID 26752291, CID 975656) is the 1st chemotype observed to modulate epigenetics in the LDR screen. It is >= 4 fold selective when compared to an AP1 signaling assay (which showed inactivity at 38 uM).
In vitro activity - HDAC Inhibition and Promoter Methylation Assays
Summary /
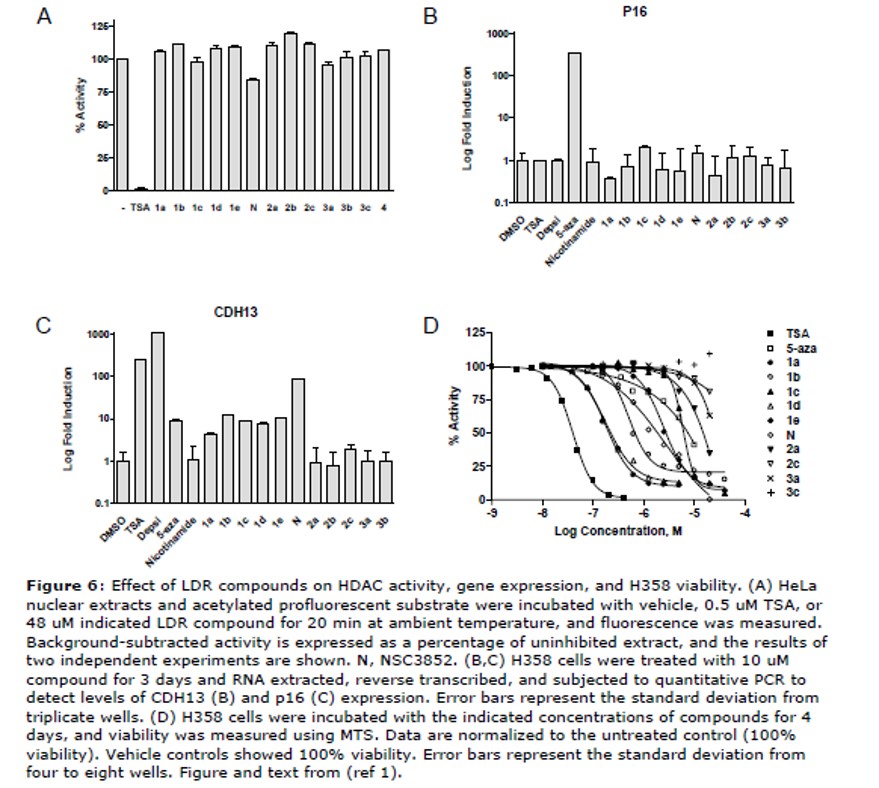
Inhibition of HDAC activity, derepression of methylated gene expression, and killing of lung cancer lines. ML065 was not observed to inhibit HDAC activity in HeLa cell extracts at 48 uM (Figure 6A). This suggested that this compound is not a general HDAC inhibitors, but rather may target different epigenetic enzymes or specific HDACs of low abundance in HeLa or LDR extracts.
Reactivation of the expression of endogenous genes silenced by promoter methylation. Human non-small cell lung cancer H358 cells harbor methylated CpG islands at the CDH13 and p16 promoters, with the latter being densely methylated and fully silenced by this modification (Phelps 1996; Virmani 2002). H358 cells were incubated with compounds for 3 days, and CDH13 and p16 transcript levels were measured by real time quantitative RT PCR. The HDAC inhibitors, depsipeptide and TSA, and the DNMT inhibitor, 5-azadeoxycytidine, reversed CDH13 silencing by 10 to 1000 fold, while only 5-azadeoxycytidine reactivated p16 expression (Figure 6B and 6C). Nicotinamide, an inhibitor of sirtuins (Bitterman 2002), and Series 2 (2a = ML065) and 3 (3a = ML064) compounds did not derepress either gene. Like depsipeptide and TSA, Series 1 compounds induced CDH13 but not p16 gene expression. Of this series, NSC3852 was the most potent, inducing expression 85 fold over basal levels while the others induced expression by 4 to 12 fold. These results indicate the LDR actives do not behave as DNMT inhibitors to derepress transcription of both p16 and CDH13 genes.
In vitro activity - Selectivity against non-small cell lung cancer
Summary /
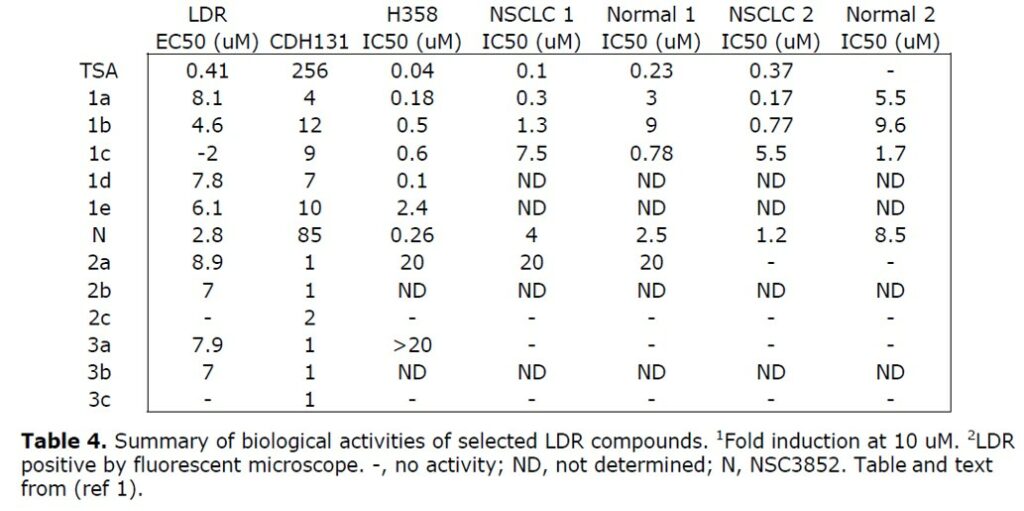
Selective against tumor cells: two non-small cell lung cancer lines and their matched normal bronchial epithelial cells, both derived from patient samples. After four days of treatment, TSA killed both NSCLC lines and one normal line with 0.1-0.4 uM IC50, but did not reduce the viability of normal line 2 at concentrations up to 0.4 uM (Figure 6D). One active and one inactive compound were tested from Series 2 and 3, all of which showed little to no activity (Table 4). For one matched set, 2a (ML065) decreased viability in both tumor and normal lines by about 50% at 20 uM. Of the three series 1 compounds tested, 1a (ML066) and 1b were potent and selective for both tumor lines (Figure 7, Table 4). ML066 was 10- to 20-fold selective for the tumor lines with an IC50 between 0.2-0.3 uM, while 1b was 7- to 12-fold selective with an IC50 of about 1 uM. 1c displayed 3- to 10-fold selectivity for the normal lines with a potency of 1-2 uM.
References
- Quantitative High-Throughput Screen for Epigenetic Modulators: Summary
- Johnson, R. L., W. Huang, et al. (2008). "A quantitative high-throughput screen identifies potential epigenetic modulators of gene expression." Anal Biochem 375(2): 237-48
- Bitterman, K. J., R. M. Anderson, et al. (2002). "Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1." J Biol Chem 277(47): 45099-107
- Virmani, A. K., J. A. Tsou, et al. (2002). "Hierarchical clustering of lung cancer cell lines using DNA methylation markers." Cancer Epidemiol Biomarkers Prev 11(3): 291-7
- Phelps, R. M., B. E. Johnson, et al. (1996). "NCI-Navy Medical Oncology Branch cell line data base." J Cell Biochem Suppl 24: 32-91