
ML251 : TbPFK (T. brucei ATP-dependent 6-phosphofructokinase) Inhibitor
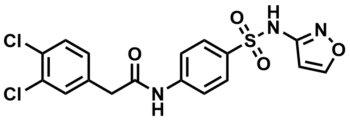
ML251
Target Name
T. brucei ATP-dependent 6-phosphofructokinase
Target Alias
TbPFK
Target Class
Transferase
Mechanism of Action
Inhibitor of TbPFK
Biological / Disease Relevance
Glycolytic Pathway, Glycolysis, Human African Trypanosomiasis (HAT)
In vitro activity
T. brucei PFK bioassay (IC50)Cellular activity
PFK secondary assay (IC50)Target Information
The protist Trypansoma brucei is the causative agent of Human African Trypanosomiasis (HAT), a disease that is endemic to sub-Saharan Africa and is responsible for approximately 50,000 deaths each year worldwide. Current therapies employed to treat HAT generally suffer from poor selectivity profiles, and consequently, lead to high rates of deleterious side effects. Furthermore, growing resistance to these drugs has highlighted a need for the identification of novel therapies. The glycolytic enzyme phosphofructokinase (PFK) has been recognized as a potential therapeutic target in the fight against the bloodstream form of T. brucei due to the reliance of this parasite on the metabolism of glucose as its sole mechanism for the generation of adenosine triphosphates (ATP). The importance of this enzyme has been confirmed by genetic validation, and thus, its inhibition may represent a novel strategy for the treatment of HAT. Currently, no inhibitors of Tb PFK have been described that possess ideal drug-like qualities or submicromolar potency in either enzymatic or live parasite culture assays. As a result, in collaboration with Professor Malcolm Walkinshaw of the University of Edinburgh, a quantitative High Throughput Screening (qHTS) campaign was initiated at the NIH Chemical Genomics Center to identify novel small molecule inhibitors of Tb PFK. The primary screen utilized a firefly luciferase-coupled assay specifically designed to monitor the production of ADP in the kinase reaction. This work, together with subsequent medicinal chemistry efforts, has identified a novel small molecule inhibitor (ML251) of Tb PFK, which is described herein to possess potent activity in enzymatic assays (IC50 = 370 nM) and toxicity against cultured T. brucei (ED50 = 16.3 uM). This compound has been subjected to a panel of in vitro absorption, distribution, metabolism, and excretion (ADME) assays and delivers a promising profile in this respect. Finally, cytotoxicity evaluations against MRC-5 human lung fibroblast cells and KB-3-1 HeLa cells, in addition to assessment against purified Bacillus stearothermophilus PFK, indicate that ML251 displays a high degree of selectivity for the targeted enzyme.
Properties
ML251
NCGC00244858
| Physical & chemical properties | ||||
|---|---|---|---|---|
| Molecular Weight | 426.3 g/mol | |||
| Molecular Formula | C17H13Cl2N3O4S | |||
| cLogP | 3.3 | |||
| PSA | 110 Ų | |||
| Storage | ||||
| Solubility | ||||
| CAS Number | ||||
SMILES:
ClC1=C(Cl)C=C(CC(NC2=CC=C(S(=O)(NC3=NOC=C3)=O)C=C2)=O)C=C1
InChI:
1S/C17H13Cl2N3O4S/c18-14-6-1-11(9-15(14)19)10-17(23)20-12-2-4-13(5-3-12)27(24,25)22-16-7-8-26-21-16/h1-9H,10H2,(H,20,23)(H,21,22)
InChIKey:
AIDVIFPYWYKRCE-UHFFFAOYSA-N
Activity
Summary activity statement /
The para-amidosulfonamide ML251 (SID 124391163; CID 53255421) represents the first small molecule to possess submicromolar inhibitory activity against Trypanosoma brucei phosphofructokinase (Tb PFK). As a molecular probe, ML251 will be invaluable for advancing our understanding of the role that glycolysis plays in the survival of members of the trypanosomatid family. Furthermore, ML251 has shown promising activity in cultured parasite growth assays. Coupled with data suggesting ML251 does not inhibit bacterial isoforms of PFK and lacks cytotoxicity in a number of human cell lines, this result provides the first evidence that specific inhibition of Tb PFK by a small molecule may be realized. Lastly, ML251 has performed extremely well in a myriad of in vitro ADME assessment assays, which provide strong support for ML251 as an excellent lead compound for further development as a small molecule chemotherapeutic for the treatment of T. brucei infection.
In vitro and cellular activity - Selectivity and Cytotoxicity Assay
| Bioassay | ML251 (IC50) |
|---|---|
|
Tb PFK |
0. 37 uM |
|
KB-3-1 cells (Cytotoxicity) |
> 46 uM |
|
PFK ADP-glo (Secondary assay) |
0.23 uM |
Summary /
ML251 is observed to be a potent inhibitor against the Tb PFK with no apparent cytotoxic effect observed (against KB-3-1). In an effort to probe the selectivity of the series for Tb PFK, all analogs were evaluated for inhibitory activity against the PFK isoform of Trypanosoma cruzi, a related trypanosomatid species responsible for Chagas disease, as well as that of Bacillus stearothermophilus, a thermophilic species of bacteria. Not surprisingly, comparable inhibitory activity against T. brucei PFK and T. cruzi PFK was observed for all compounds, as these isoforms display a high degree of similarity, with greater than 90% sequence identity. Cross-species activity of small molecules across multiple members of the trypanosomatid family is a well-recognized phenomenon, and given the tremendous burden both T. brucei and T. cruzi place on global health, this shared inhibition of PFK would not be considered a liability for the lead series. Encouragingly, when the same compounds were tested against the B. stearothermophilus isoform of PFK, no inhibitory activity was observed (PFK isoform selectivity data for ML251 given in Figure 2). As the Bacillus isoform shares only 30% sequence identity with T. brucei, this lack of inhibitory activity seems relatively unsurprising. Following these studies, all analogs were evaluated for general cytotoxicity against MRC-5 human lung fibroblast and KB-3-1 human cervical carcinoma cell lines, as purified human PFK is not available. These studies revealed essentially no cytotoxicity for the lead series, providing further evidence of its selectivity for trypanosomatid isoforms of PFK (cytotoxicity data for ML251 is given in Figure 1).
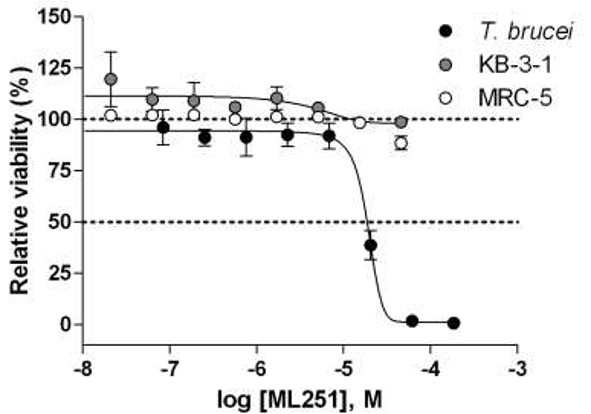
Figure 1. Cytotoxicity of ML251 against cultured T. brucei brucei (black circles), KB-3-1 human cervical carcinoma (grey circles) and MRC-5 human lung fibroblast (white circles) cell lines after 72 hr incubation. Each point represents the mean±SD for three independent experiments.
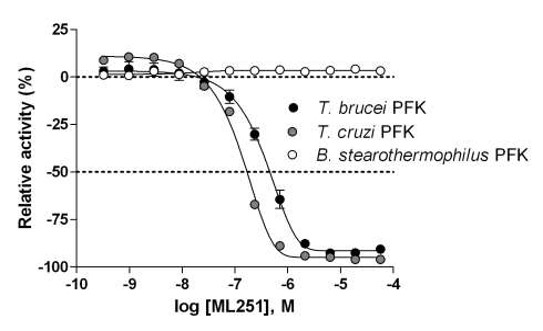
Figure 2. Inhibitory activity of ML251 against T. cruzi (grey circles), T. brucei (black circles), and B. stearothermophilus (white circles) isoforms of PFK.
In vitro activity - ADME profiling
Summary /
ML251 showed promising ADME profile (Table 1).
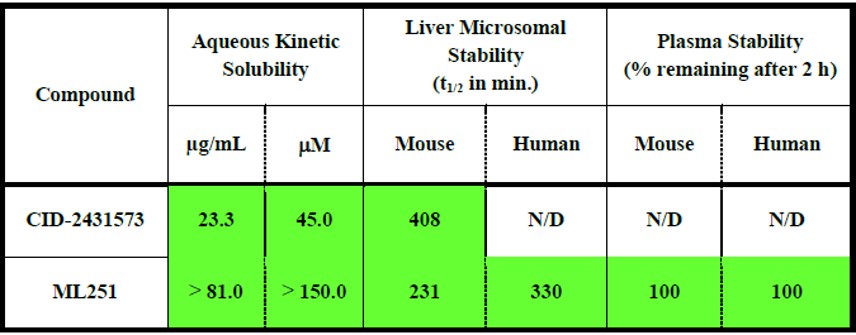
Table 1. ADME profile for qHTS hit 1 (CID-2431573) and 30 (ML251).
In vitro activity - Molecular modeling: compound docking
Summary /
While a number of analogs had been generated in the first two rounds of synthesis that possessed comparable activity to the original hit 1, failure to improve upon this potency led to the decision to utilize the previously solved ATP-bound crystal structure of Tb PFK as a basis for molecular modeling efforts to guide future rounds of analog development. Thermal melt data suggested that 1 engages in competition for binding with F6P, as stabilization of Tb PFK is observed in the presence of ATP and inhibitor, but not in the presence of F6P and inhibitor. This finding led to docking of 1 in the sugar binding site and conformational energy minimization delivered a binding orientation in which the 3,4- dichlorobenzyl functionality of 1 extends into a hydrophobic pocket generated by the Leu346, Ile347, and Ile349 residues of Tb PFK. Important hydrogen bonding interactions are also predicted in this rendering for the sulfonamide linker with the Arg383 residue. Lastly, the modeling predicts occupation of the 5-methyl-3-aminoisoxazole functionality within a highly hydrophilic region of the Tb PFK active site. It is within this site that the LYS226 residue is predicted to engage in a hydrogen bond with the nitrogen center of the isoxazole functionality of 1. This suggested mode of binding was further corroborated by mechanistic enzyme studies that were performed on ML251.
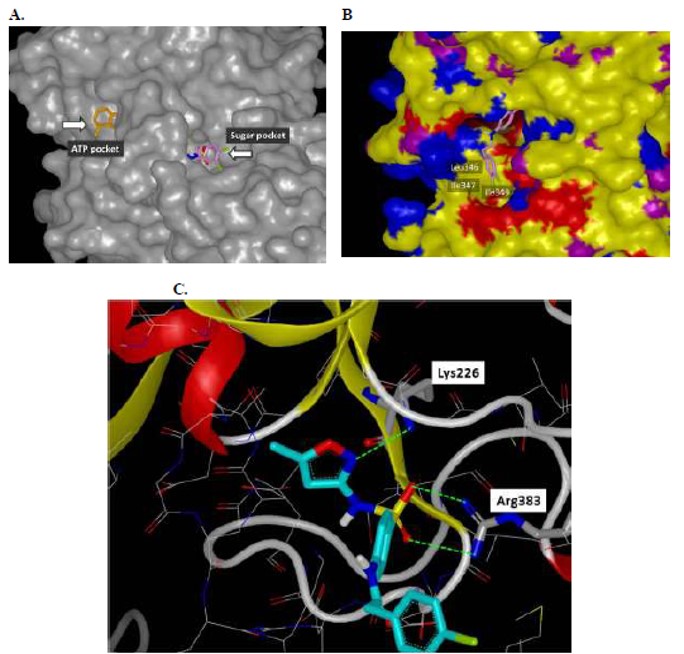
Figure 3. Modeling of CID-2431573 in F6P binding site of Tb PFK. A) Rendering of the tunnel-like active site of Tb PFK and docking of CID-2431573 (pink) in the F6P binding site (ATP is shown in gold); B) Alternative view showing hydrophobic pocket that is predicted to be occupied by the dichlorobenzyl functionality of CID-2431573 (pink); C) Interior of Tb PFK active site and predicted interaction with CID-2431573 (cyan).
In vitro activity - Mechanism of action
Summary /
Inclusion of saturating concentrations of ATP or F6P in the ADP-Glo inhibition assay showed a marked right-shift in ML251 potency under high F6P conditions (Figure 13, Panel A); the IC50 of ML251 against Tb PFK shifted from 365 nM under standard conditions to 2.06 μM in the presence of saturating F6P, demonstrating a 5.6-fold shift in potency. Inclusion of saturating ATP, by contrast, demonstrated virtually no change in ML251 potency, with an IC50 of 326 nM. These observations were followed up with a mechanistic competition study in which individual substrates and ML251 were titrated in a coupled PFK assay system. ML251 demonstrated a minimal effect on the Km of ATP, yet appeared to affect Vmax (Figure 13, Panel B, transformed to Lineweaver-Burk plot for ease of visualization), suggesting that it is noncompetitive with respect to ATP. F6P, conversely, showed no change in Vmax in the presence of ML251, but did show a shift in Km, suggesting that the compound may be competitive with respect to F6P (Figure 13, Panel C). These observations agree with the results of both our biochemical assays and molecular modeling in suggesting that the lead chemical series preferentially binds in the F6P region of the Tb PFK active site.
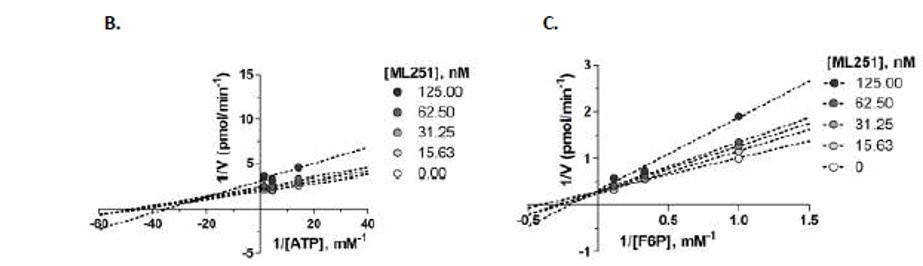
Figure 4. Evaluation of substrate competition of ML251 with F6P and ATP in Tb PFK. A. Inhibitory activity of ML251 in ADP-Glo assay under standard (white circles), saturating ATP (light grey circles) and saturating F6P (dark grey circles) conditions; B. Lineweaver-Burk plot of competition relationship between ML251 and ATP; C. Lineweaver Burk plot of competition relationship between ML251 and F6P.
References
- PubChem link: qHTS Assay to Find Inhibitors of T. brucei Phosphofructokinase: Summary
- Walsh MJ, Brimacombe KR, Vásquez-Valdivieso MG, et al. Identification of Selective Inhibitors of Phosphofructokinase as Lead Compounds Against Trypanosomiasis. 2011 Oct 18 [Updated 2013 Feb 25]. In: Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK154498/