
ML247 : GAA (Lysosomal alpha-glucosidase) Modulator
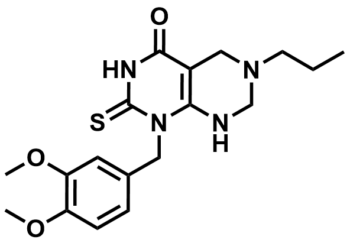
ML247
Target Name
Lysosomal alpha-glucosidase
Target Alias
GAA
Target Class
Glucosidase
Mechanism of Action
Modulator of GAA
Biological / Disease Relevance
Glycogen Storage Disease II, Pompe Disease, Lysosomal Storage Disorder (LSD),
In vitro activity
Acid alpha-glucosidase bioassay (IC50)Cellular activity
Lysosomal GAA translocation (Patient fibroblast)Target Information
Glycogen storage disease II, or Pompe disease, is a rare and often fatal autosomal recessive lysosomal storage disorder (LSD) caused by the dysfunction of the lysosomal enzyme acid alpha-glucosidase (GAA). Accumulation of GAA’s substrate, glycogen, causes enlargement of cellular lysosomes, adversely affecting many cells, especially heart and skeletal muscle tissues. The only FDA approved treatment for Pompe disease is enzyme replacement therapy, called Myozyme, which has significant limitations. Importantly, of the over 100 different mutations known to cause Pompe disease, many retain enzymatic activity in vitro, although the structural changes induced by mutants affect trafficking of the enzyme to the lysosome. Small molecule chaperones can be used to correct this trafficking defect. These compounds bind to the protein in the endoplasmic reticulum, accelerating the folding process and increasing their translocation to the lysosome, thereby reducing substrate accumulation. Several iminosugar inhibitors of GAA, such duvoglustat, are known to chaperone the translocation of mutant GAA proteins. However, their impact on substrate reduction may be limited by their continued inhibition of the target enzyme, as well as limited selectivity towards GAA. We have previously reported an inhibitor (ML201) and through further work, we have now also identified the first non-inhibitory small molecule chaperone of acid alpha glucosidase, ML247. Here, we demonstrate that ML247 enhances mutant enzyme translocation using Pompe patient-derived fibroblasts. ML247 displays reasonable pharmacokinetics and might serve as a pivotal first step in efforts to develop a non-inhibitory molecular chaperone for the treatment of Pompe disease.
Properties
ML247
NCGC00183885
| Physical & chemical properties | ||||
|---|---|---|---|---|
| Molecular Weight | 376.5 g/mol | |||
| Molecular Formula | C18H24N4O3S | |||
| cLogP | 2 | |||
| PSA | 98.2 Ų | |||
| Storage | ||||
| Solubility | ||||
| CAS Number | ||||
SMILES:
CCCN1CNC2=C(C(NC(N2CC3=CC(OC)=C(OC)C=C3)=S)=O)C1
InChI:
1S/C18H24N4O3S/c1-4-7-21-10-13-16(19-11-21)22(18(26)20-17(13)23)9-12-5-6-14(24-2)15(8-12)25-3/h5-6,8,19H,4,7,9-11H2,1-3H3,(H,20,23,26)
InChIKey:
FBESZLCTQRAXBF-UHFFFAOYSA-N
Activity
Summary activity statement /
Mutations in the lysosomal enzyme acid alpha-glucosidase (GAA) are the underlying cause of Pompe disease. Here, we present a small molecule chaperone that can correct the misfolding and mistrafficking of disease-causing mutant protein. All previously described small molecule chaperones of GAA are inhibitors of the enzyme. Here, we disclose a new class of non-inhibitory, selective small-molecule GAA chaperones ML247 (SID 85267344; CID 44246403) and analogs that increase the translocation of several GAA mutants in primary patient fibroblasts without inhibiting the translocated protein’s specific activity. These molecules increase the rate of hydrolysis in enzymatic assays, measured through several of the classical fluorescent substrates of GAA. ML247 displays reasonable PK properties and exposure in vivo and is an ideal candidate for further characterization of its ability to reduce substrate accumulation in vivo.
In vitro activity - Selectivity and Cytotoxicity Assay
| Bioassay | ML247 (IC50) |
|---|---|
|
Acid alpha-glucosidase |
2.818 uM |
|
beta-glucosidase (Anti-Target) |
> 57 uM |
|
alpha-galactosidase (Anti-Target) |
> 57 uM |
Summary /
ML247 is found to have > 10 fold selective against the GAA vs. other metabolic enzymes b-glucosidase and a-galactosidase.
Cellular Activity - Functional assay: Chaperone Activity in Pompe Patient-derived Fibroblasts
Summary /
To measure the capacity of our compounds to activate ER-lysosome translocation of GAA, we treated Pompe patient-derived fibroblasts with compound for five days, followed by immunostaining (GAA and Cathepsin D as lysosomal marker), wash, and image analysis (Parenti 2007, Okumiya 2007). Pompe fibroblasts are difficult to obtain, and in general, they grow slowly; they can only be expanded by a few passages. Two cell cultures derived from a single patient with the mutations p.Y455C/p.G638W were obtained. The residual specific activity of these patient-derived fibroblasts is around 3% of the activity displayed by fibroblast with wildtype GAA. Although the specific activity of GAA in fibroblasts (skin biopsies) is definitive for a Pompe disease diagnosis, fibroblasts do not express GAA in abundant amounts, and that expression decreases with increasing passage number, even for wildtype cells. Nevertheless, the present patient-derived cells had sufficient protein expression to enable us to characterize the chaperone activity of this series, which is done by examining the co-localization of GAA staining with lysosome marker staining (CathD). No lysosomal GAA staining can be seen in DMOS vehicle treated Pompe cells. As a reference, in our hands, the GAA signal in wildtype fibroblast by passage 8 could be observed by staining in 10% of the cells. The inhibitory chaperone duvoglustat is a non-selective iminosugar currently in clinical trials for Pompe disease. The chaperone activity of duvoglustat varies by mutation, and there is no previous literature indicating whether duvoglustat is active against p.Y455C/p.G638W GAA mutations. No chaperone activity was observed at compound concentrations of 1, 5, and 15 μM. At 20 μM, translocation of GAA to the lysosome occurred in 3% of the cells. These values are in the same range as those reported for other GAA mutants, where duvoglustat increased GAA translocation at 20 μM. Duvoglustat does not increase the translocation of wildtype GAA. In contrast, probe molecule ML 247 and some of its analogs increases translocation of GAA to lysosomes in patient-derived fibroblasts. ML247 gave the most robust responses, with around 10% translocation of mutant GAA to the lysosomes at 20 μM. Translocation could still be observed at 15 and 10 μM, but not at 5 μM (Figure 1). In addition, profound restoration of lysosomal GAAA in patient-derived fibroblast is observed after 5 day incubation with 20 uM ML247 (Figure 2). Comparison between the results obtained with duvoglustat vs. ML247 show that at the same concentration, the probe molecule is superior than the prior art molecule duvoglustat.
Figure 1. Probe compound ML247 restores GAA translocation to the lysosome (blue arrows). False-colored images of fibroblast cells show DAPI nuclear stain (blue), the lysosomal marker Cathepsin D (red) and GAA (green) in cells from (A) an unaffected donor with wildtype GAA, and (B-E) Pompe patient-derived cells with p.Y455C/p.G638W mutations. Cells were cultured for five days with compound that was dispensed using a DMSO vehicle. Yellow color (overlapping green and red) indicates co-localized lysosomal GAA protein, typically present in (A) DMSO vehicle treated wildtype cells, but absent in (B) DMSO vehicle treated patient-derived cells, and partially restored with (C) 20 μM duvoglustat, (D) 15μM compound 1, and more strongly with (E) 15 μM ML247.
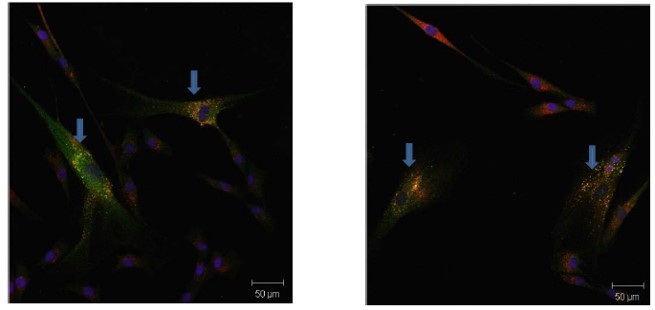
Figure 2. Two representative fields showing profound restoration of lysosomal GAA in patient-derived fibroblasts (blue arrows), upon five day incubation with 20 μM probe compound ML247. Coloring scheme is the same as Figure 1.
In vitro and vivo activity - ADME and PK profiling
Summary /
ML247, has excellent Caco-2 permeability (> 5cm-8/s) and robust stability, with >60% remaining after a 60 minute incubation with mouse liver microsomes. The addition of NADPH also had little effect on compound stability, indicating only very modest P450 mediated metabolism (Table 1). ML247 was further studied in a single-dose pharmacokinetics experiment. At 50 mg/kg IP dosing, no acute toxicity was observed (Figure 3). Compound half-life was 2.75 hours in plasma, with concentrations in plasma, liver and heart all exceeding 10 μM at one hour. Pharmacokinetic properties and therapeutic window considerations place ML247 as an excellent candidate for further advancement studies toward in vivo efficacy model and eventually in the treatment of Pompe disease.
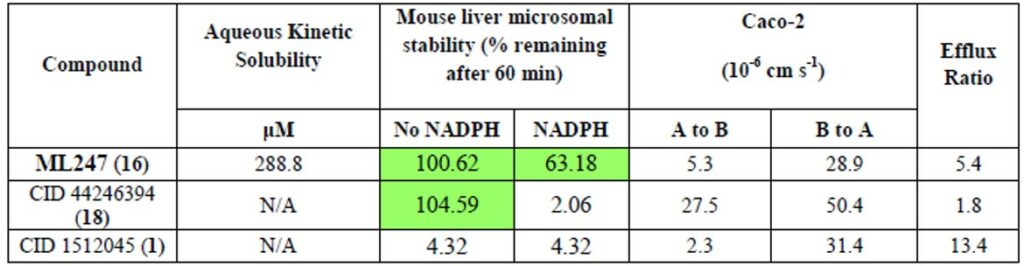
Table 1. In vitro ADME profile of ML247.
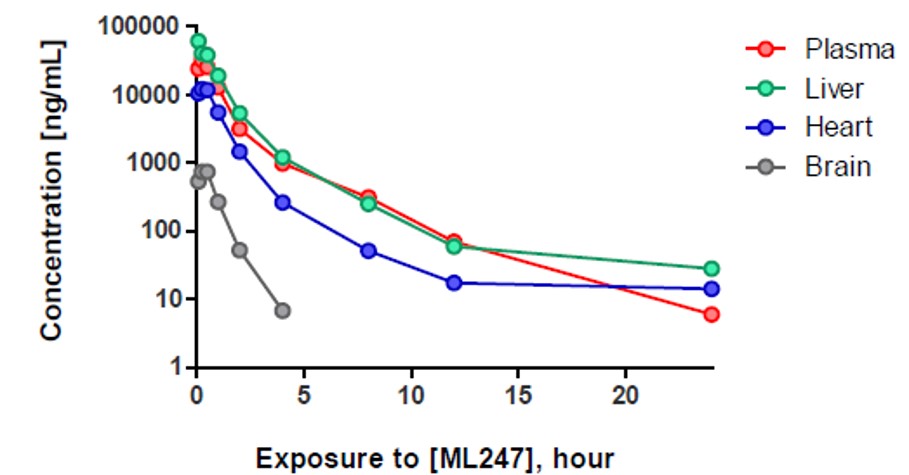
Figure 3. In vivo exposure by ML247 following a single 50 mg/kg IP dose in mice.
References
- PubChem link: Quantitative High-Throughput Screen for Inhibitors and Activators of Human alpha-Glucosidase as a Potential Chaperone Treatment of Pompe Disease: Summary
- Marugan JJ, Zheng W, Ferrer M, et al. Discovery, SAR, and Biological Evaluation of a Non-Inhibitory Chaperone for Acid Alpha Glucosidase. 2011 Dec 16 [Updated 2013 May 3]. In: Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK153221/
- Parenti G, Zuppaldi A, Gabriela Pittis M, et al. Pharmacological enhancement of mutated alpha-glucosidase activity in fibroblasts from patients with Pompe disease. Mol Ther. 2007;15(3):508-514. doi:10.1038/sj.mt.6300074
- Okumiya T, Kroos MA, Vliet LV, Takeuchi H, Van der Ploeg AT, Reuser AJ. Chemical chaperones improve transport and enhance stability of mutant alpha-glucosidases in glycogen storage disease type II. Mol Genet Metab. 2007;90(1):49-57. doi:10.1016/j.ymgme.2006.09.010