
ML168 : HTT (Huntingtin) Inhibitor
ML168
Target Name
Huntingtin
Target Alias
HTT
Target Class
Microtubule-mediated Transport
Mechanism of Action
Inhibitor of HTT
Biological / Disease Relevance
Huntington's disease, Neurodegenerative disease
In vitro activity
Q103 HTT induced cytotoxicity bioassay (IC50)Cellular activity
HTTQ103 serum-deprived striatal cell cytoprotection assayTarget Information
Huntington’s disease is a progressive, neurodegenerative disorder whose genetic cause is an expansion of a CAG trinucleotide repeat in exon 1 of the Huntington gene that results in a long chain polyglutamine (polyQ) tract (MacDonald 1993). Clinical and statistical analysis have shown that the number of polyQ repeats correlates with the probability of developing the disease, with patients that have greater than 36 to 40 polyQ having a high probability of developing this disorder (Bates 2003, Goehler 2004). Although the function of Huntingtin protein is complex and not completely understood, it is known that polyQ repetitions in the htt protein increase the propensity for aggregate formation. The nature of these aggregates, as to whether they are cytoprotective, cytotoxic, or a combination of both, is highly debated in the field (Rubinsztein 2003, walker 2007). It is also known that polyQ repetitions trigger a not-well understood apoptotic process that ultimately results in neurodegeneration. A PC12 cell line stably harboring an ecdysone inducible fusion of the of mutant Huntingtin protein (htt exon 1 containing a 103 poly Q repeat, called Q103HTT) to GFP was used as the cell line for HTS (Aiken 2004). Induction of the fusion protein by tebufenozide resulted in the formation of GFP aggregates and increased cytotoxicity. At the NCGC, we developed a cell-based, multiplexed high-throughput screening assay that allows us to quantify the effect of small molecules on Huntingtin protein aggregation and cytotoxicity. In this report, we present our initial SAR evaluation of one of our active lead series. In addition, we also report our initial findings regarding its potential mechanism of action.
Properties
ML168
NCGC00037991
| Physical & chemical properties | ||||
|---|---|---|---|---|
| Molecular Weight | 326.4 g/mol | |||
| Molecular Formula | C14H10N6S2 | |||
| cLogP | 3.42707 | |||
| PSA | 65.04 Ų | |||
| Storage | ||||
| Solubility | 10 mM in DMSO | |||
| CAS Number | ||||
SMILES:
CN1N=NN=C1SC2=C3C(C4=CC=CC=C4)=CSC3=NC=N2
InChI:
1S/C14H10N6S2/c1-20-14(17-18-19-20)22-13-11-10(9-5-3-2-4-6-9)7-21-12(11)15-8-16-13/h2-8H,1H3
InChIKey:
HIPMWEIBNDUPSH-UHFFFAOYSA-N
Activity
Summary activity statement /
ML168 (CID 2432214, SID 89650053 ), which is able to protect cells from cytotoxicity induced by the expression of a large mutant poly-Q expansion Huntingtin (HTT) protein by inhibiting the activation of the intrinsic apoptotic pathway. This probe can be used in cellular assays and ex vivo experiments to study and inhibit the initiation of apoptosis induced by the expression of mutant, cytotoxic HTT protein. This probe will be of great interest to the scientific community that studies Huntington’s disease and other neurodegenerative diseases.
In vitro activity - Selectivity and Cytotoxicity Assay
| Bioassay | (IC50) |
|---|---|
|
Q103HTT induced cytotoxicity |
7.94 uM |
|
Q25HTT induced cytotoxicity (Anti-Target) |
>92 uM |
|
HTTQ103 serumdeprived striatal cell cytoprotection |
Cytoprotective |
Summary /
ML168 showed Q103HTT cytoprotective effect when assayed. It reversed the stress-induced apoptosis specifically in the cells expressing mutant Huntingtin protein (Q103HTT) compared to the cells expressing wildtype (Q25HTT), indicating a selective reversal of the Huntington phenotype with these cells.
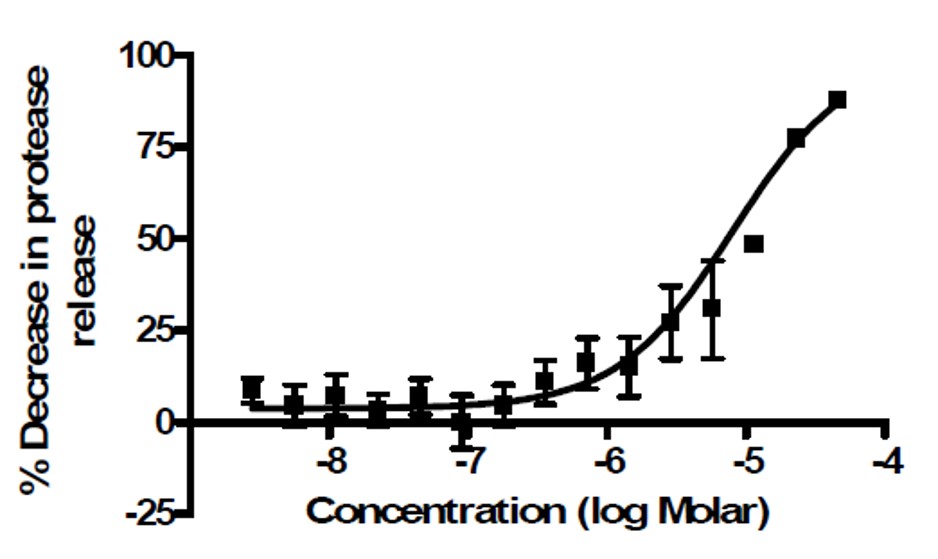
Figure 1: Activity of ML168 (MLS000089318, CID 2432214) in preventing Q103HTT induced cytotoxicity as measured by the protease release assay.
Cellular activity - Striatal cell assay
Summary /
Striatal cells are of particular relevance to the pathology of Huntington’s disease. The phenotype observed with expression of the mutant Huntingtin protein is an enhanced sensitivity to cellular stress, in this case being serum deprived culture. For this experiment, striatal cells with wildtype (Q7/Q7) and mutant full-length protein (Q111/Q111) were grown without FBS and treated with the 2 probes of interest. Both compounds reversed the stress-induced apoptosis specifically in the cells expressing mutant Huntingtin protein, indicating a selective reversal of the Huntington phenotype with these cells.
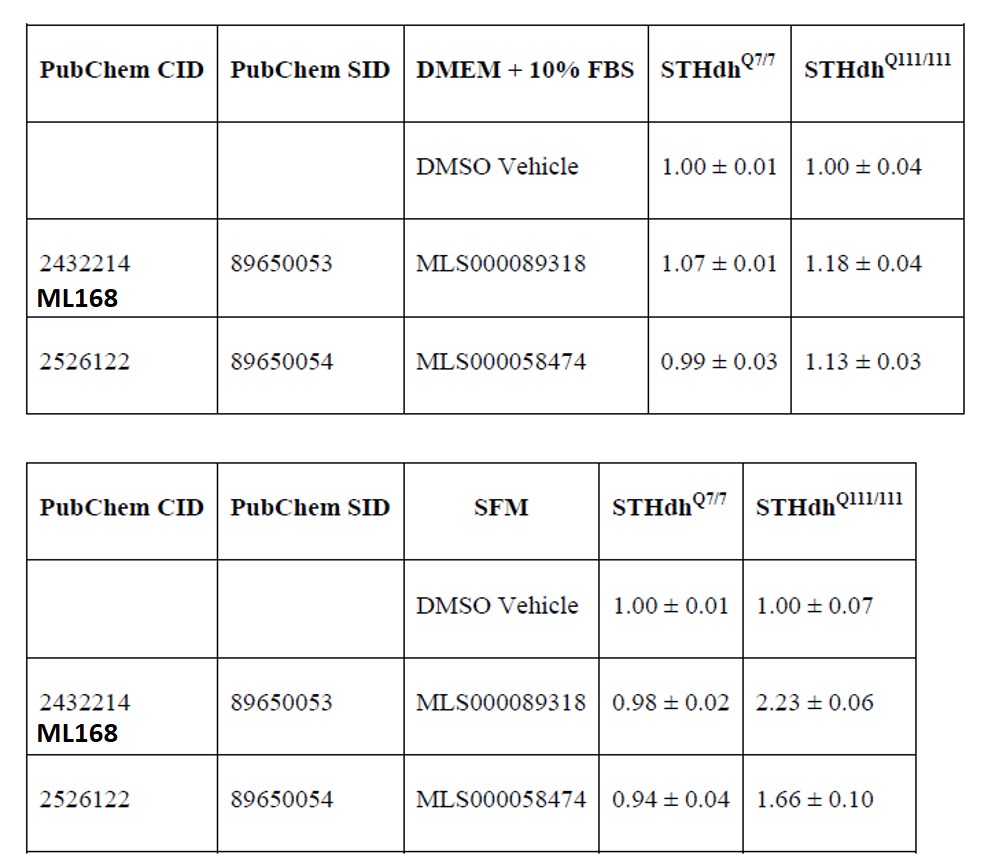
Table 1. ATP quantitation of wild type and mutant Huntington striatal cells in normal growth media (DMEM + 10% FBS) and serum deprived media after compound treatment. Data are expressed as the AVG ± STDV of the ratio of the treatment to DMSO (vehicle) control for that cell type and media condition. ML168 (MLS000089318) and MLS000058474 prevent cytotoxicity induced by serum deprivation in cells expressing the mutant huntingtin protein.
Cellular activity - Mechanism of Action Studies
Summary /
ML168 (MLS000089318, CID 2432214) is able to block the activation of caspase 9 and its downstream target, caspase 3. Surprisingly, it prevents activation of apoptotic caspases in both wild type and mutant Huntington striatal cells under serum deprivation conditions. Caspases 9 and 3 are involved in the initiation of the intrinsic apoptotic pathway. It has been extensively reported that the apoptotic intrinsic pathway is activated in Huntington’s disease, ALS, Niemann-Pick disease type C, Lysosomal Cell Death, Ischemia, viral and bacterial infection, and ceramide induced neuronal death. Thus, the inhibition of this intrinsic apoptotic pathway looks to be the mechanism of action of this probe in the cell-based Huntington model.
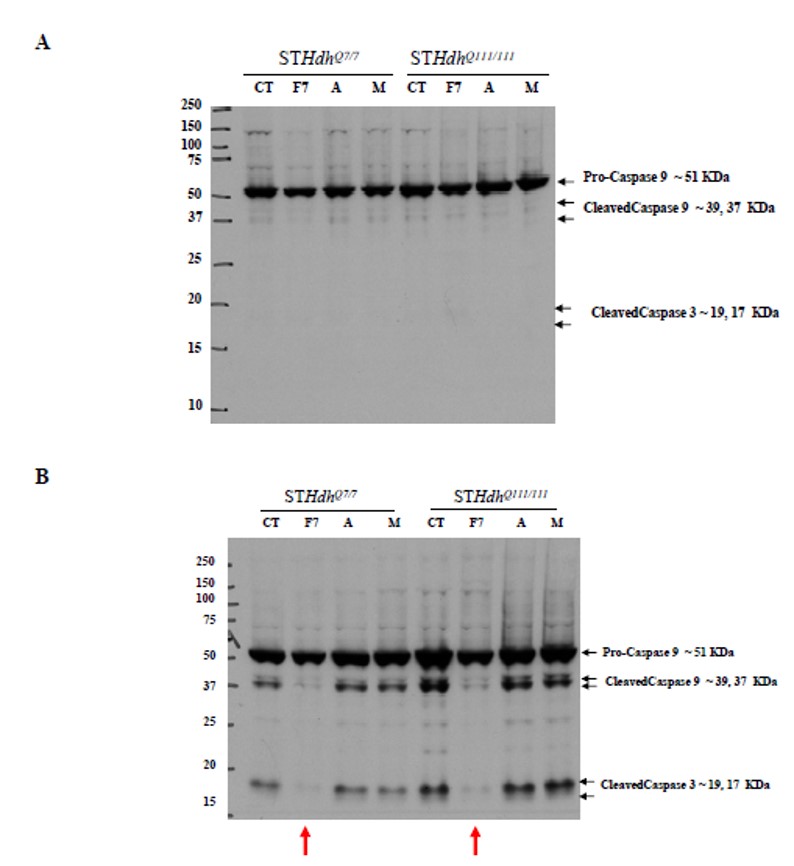
Figure 2. Western blot of caspase protein following compound treatment. ML168 (MLS000089318) protects the cells from serum deprived apoptotic cell death by inhibiting the activation of caspase 9 and its downstream target, caspase 3. A) Caspase 9 and 3 activation after 16 hour treatment under normal growth conditions (DMEM + 10% FBS). B) Caspase 9 and 3 activation after 16 hour treatment under serum deprived growth conditions. Column labels: CT = DMSO (vehicle) treatment, F7 = ML168 (MLS000089318, CID 2432214), A and M are two other compounds that affect the Huntington cellular phenotype, whose identities were not disclosed by the assay provider. The red arrow indicates the specific activity of ML168 (MLS000089318, CID 2432214) under serum deprivation conditions.
References
- PubChem link: Quantitative High-Throughput Multiplex Assay to Identify Dual Action Probes in a Cell Model of Huntington: Summary
- Titus S, Elia N, Southall N, et al. Identification of compounds which inhibit cytotoxicity associated with mutant Huntingtin protein expression. 2010 Mar 30 [Updated 2011 Mar 11]. In: Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK56233/
- MacDonald, M.E., et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993 Mar 26;72(6):971-83. doi: 10.1016/0092-8674(93)90585-e. PMID: 8458085.
- Bates G. Huntingtin aggregation and toxicity in Huntington's disease. Lancet. 2003 May 10;361(9369):1642-4. doi: 10.1016/S0140-6736(03)13304-1. PMID: 12747895.
- Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, Droege A, Lindenberg KS, Knoblich M, Haenig C, Herbst M, Suopanki J, Scherzinger E, Abraham C, Bauer B, Hasenbank R, Fritzsche A, Ludewig AH, Büssow K, Coleman SH, Gutekunst CA, Landwehrmeyer BG, Lehrach H, Wanker EE. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington's disease. Mol Cell. 2004 Sep 24;15(6):853-65. doi: 10.1016/j.molcel.2004.09.016. Erratum in: Mol Cell. 2005 Jul 22;19(2):287. Buessow, Konrad [corrected to Büssow, Konrad]. PMID: 15383276.
- Rubinsztein DC, Carmichael J. Huntington's disease: molecular basis of neurodegeneration. Expert Rev Mol Med. 2003 Aug 22;5(20):1-21. doi: 10.1017/S1462399403006549. PMID: 14585171.
- Walker FO. Huntington's Disease. Semin Neurol. 2007 Apr;27(2):143-50. doi: 10.1055/s-2007-971176. PMID: 17390259.
- Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs to treat Huntington's disease. Neurobiol Dis. 2004 Aug;16(3):546-55. doi: 10.1016/j.nbd.2004.04.001. PMID: 15262266.