
ML321 : DRD2 (D(2) Dopamine Receptor) Antagonist
ML321
Target Name
D(2) Dopamine Receptor
Target Alias
DRD2
Target Class
G-protein Coupled Receptor
Mechanism of Action
Antagonist of DRD2
Biological / Disease Relevance
Cell Signaling Processes, Dopamine receptors (DARs), CNS diseases
In vitro activity
D2 DAR bioassay (AC50)In vitro activity
D2 binding Ki (AC50)In vitro activity
D2 beta-arrestin (AC50)Target Information
Dopamine receptors (DARs) are members of the G protein-coupled receptors (GPCRs) superfamily which play a critical role in cell signaling processes, especially modulating the transfer of information within the nervous system (Kuhar 1999). Amongst DARs, the D2 receptor is arguably one of the most validated drug targets in neurology and psychiatry (Beaulieu 2011). There is a strong correlation between the clinical doses of neuroleptics and their affinity for brain D2 receptors (Seeman 2000). Despite numerous attempts of producing D2 selective modulators, current drugs display poor selectivity between D2 and D3 DARs. This is because D2 is closest to D3 in terms of sequence homology and signaling transduction pathways hence it pose a challenge to selectively regulate the two receptors (Cho 2010, Miyake 2012). However given the success, a potent and selective D2 DAR antagonist would be of particular interest for the treatment of a variety of related CNS diseases (Ginovart 2012, Le Foll 2009). Here we present the discovery of a novel series of selective small molecule D2 DAR antagonists from a quantitative high-throughput screen (qHTS) campaign. Optimized lead compound in this series exhibits an excellent D2 versus D1 , D3 , D4 and D5 receptor selectivity. In a panel of GPCR binding assays, ML321 shows a cleaner profile compared to the best previously reported selective D2 DAR antagonist. Furthermore, ML321 showed good in vitro ADME data and in vivo pharmacokinetic (PK) properties. We therefore believe that this probe can be a very useful pharmacological tool to perform proof-of-concept studies in animal models and may be an ideal starting point for further development into drug-like molecules for the treatment of a variety of CNS diseases including Tourette’s syndrome, tardive dyskinesia, dystonia, Huntington’s chorea, and especially schizophrenia.
Properties
ML321
NCGC00370678-03
| Physical & chemical properties | ||||
|---|---|---|---|---|
| Molecular Weight | 410.5 g/mol | |||
| Molecular Formula | C21H18N2O3S2 | |||
| cLogP | 2.7 | |||
| PSA | 114 Ų | |||
| Storage | ||||
| Solubility | ||||
| CAS Number | ||||
SMILES:
CN1C2=C([S@+](C3=C(C1=O)C=CC=C3)[O-])C=CC(C(NCCC4=CC=CS4)=O)=C2
InChI:
1S/C21H18N2O3S2/c1-23-17-13-14(20(24)22-11-10-15-5-4-12-27-15)8-9-19(17)28(26)18-7-3-2-6-16(18)21(23)25/h2-9,12-13H,10-11H2,1H3,(H,22,24)/t28-/m0/s1
InChIKey:
YXLLQNMKIDBOGH-NDEPHWFRSA-N
Activity
Summary activity statement /
Preferential inhibition remains to be a limitation for D2 prior art antagonists. The probe ML321 (SID 136882616; CID 57377246) demonstrates excellent D2 versus D3 DAR selectivity with a very clean selectivity profile in a panel of GPCR binding assays. The selective D2 (versus D3 ) ML321 probe will be of interest to biologist to further elucidate the role of the D2 receptors on CNS diseases. ML321 will be used by researchers as a pharmacological tool for the dissection of D2 DAR signaling pathways in vitro and in vivo in response to a variety of biological conditions. Given its reasonable ADME and pharmacokinetic properties, ML321 will also provide scientists a tool to perform proof-of-concept studies in animal model. Moreover, ML321 can be further development into drug-like molecules subsequently giving clinicians a starting point for the development of novel or improved treatments (with less side effects) for Tourette’s syndrome, tardive dyskinesia, dystonia, Huntington’s chorea, and especially schizophrenia where D2 receptor is observed to play a crucial role.
Cellular Activity - Selectivity assay
Summary /
ML321 is potent (< 1 uM AC50) and found to have > 17 – 22 fold selective against the D2 dopamine receptor (DAR) vs. other DAR.
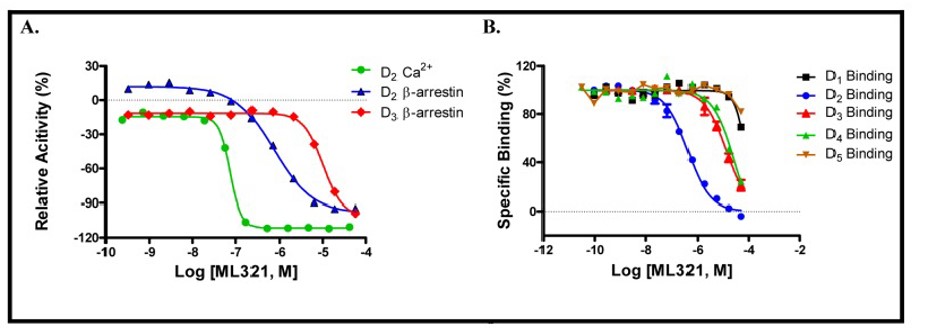
Figure 1. Dose response activity of ML321 in (A) D2 Ca++ assay (green, AC = 0.070 μM), D2 β-arrestin assay (blue, AC = 0.725 μM), and D3 β-arrestin assay (red, AC = 12.9 μM). (B) Graphical representation of the dose response curves of ML321 in binding assays for D1 (black, Ki = 67.1 μM), D2 (blue, Ki = 0.1 μM), D3 (red, Ki = 2.9 μM), D4 (green, Ki = 8.48 μM) and D5 (brown) showing preferential inhibition for the D receptor.
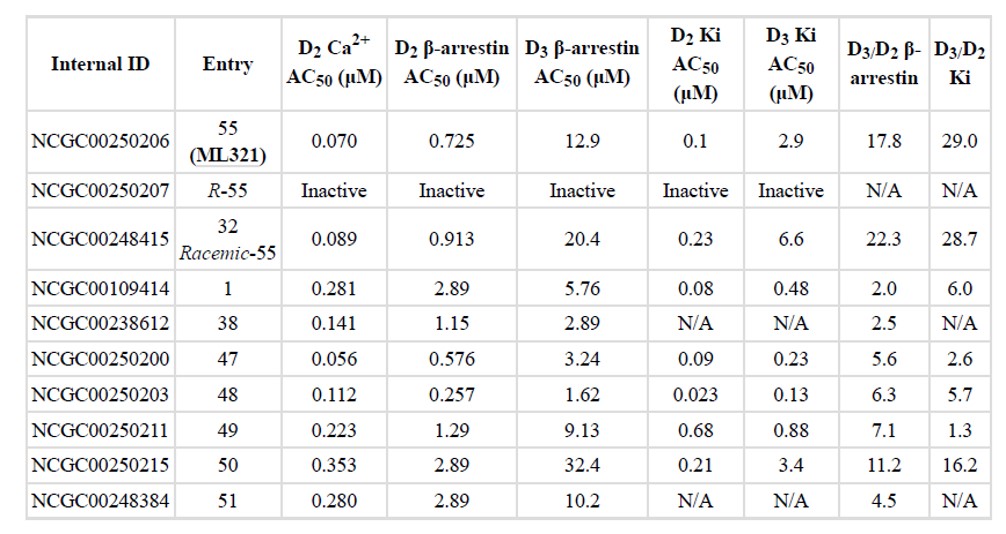
Table 1. Activity profile of the probe ML321 and relative analogs against D2 Ca++ assay in D2 expressing HK293 T-Rex™ cell line, D2 β-arrestin assay in D2 receptor PathHunter® β-arrestin cells, D3 β-arrestin assay in D3 PathHunter® β-arrestin cells, and D2 or D3 radio-ligand binding assays in HEK cell line expressing either D2L or D3 human dopamine receptor.
In vitro and vivo activity - ADME and Pharmacokinetic (PK) profile
Summary /
ADME profile for probe ML321 and a relative analog showed that the probe has good stability and permeability.
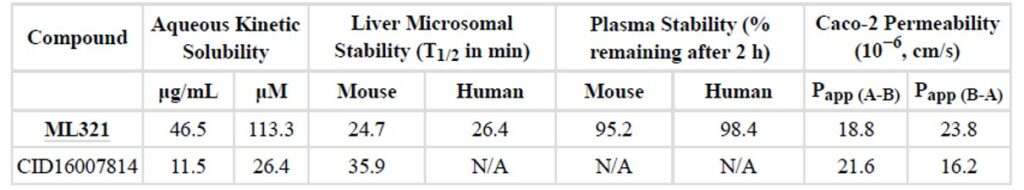
Table 2. In vitro ADME profile for probe ML321 and a relative analog NCGC00109414 (CID16007814).
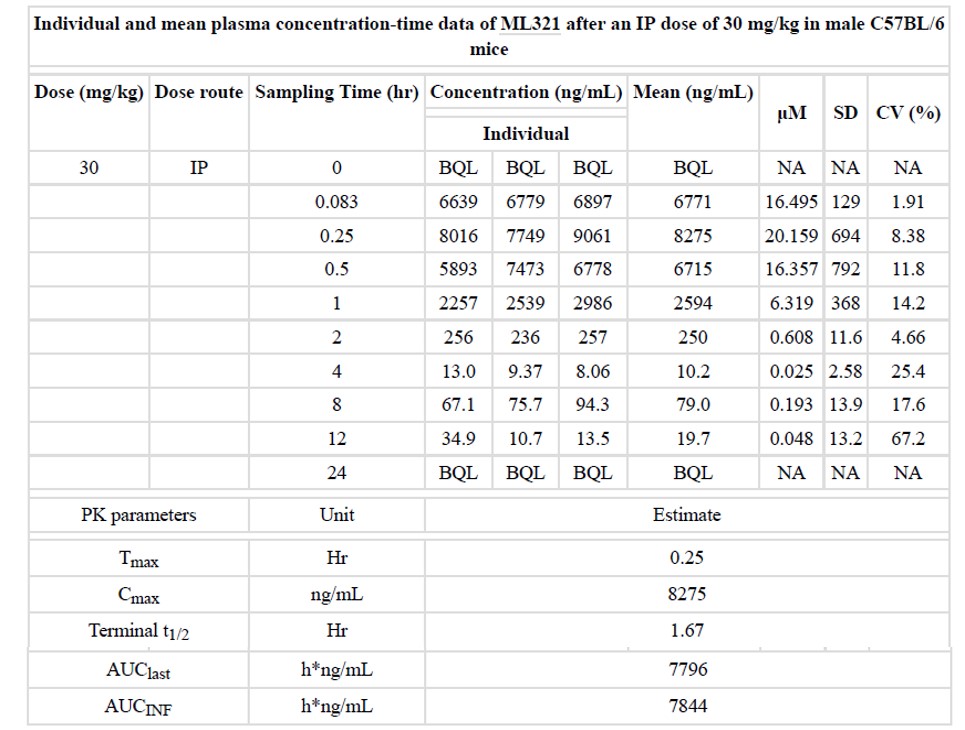
Table 3. Summary of in vivo PK data for ML321 in C57BL/6 mice (BQL = Below quantifiable limit of 1 ng/mL for ML321 in male C57BL/6 plasma. NA = Not available).
In vivo activity - Mean plasma and Brain concentration-time profile in mice
Summary /
The mice appeared less activity at 5 min post dosing, and it lasted for about 2 hr. The IP dosing solution was prepared in 10% NMP + 20% PEG400 + 70% of 25% HP-β-CD in water.
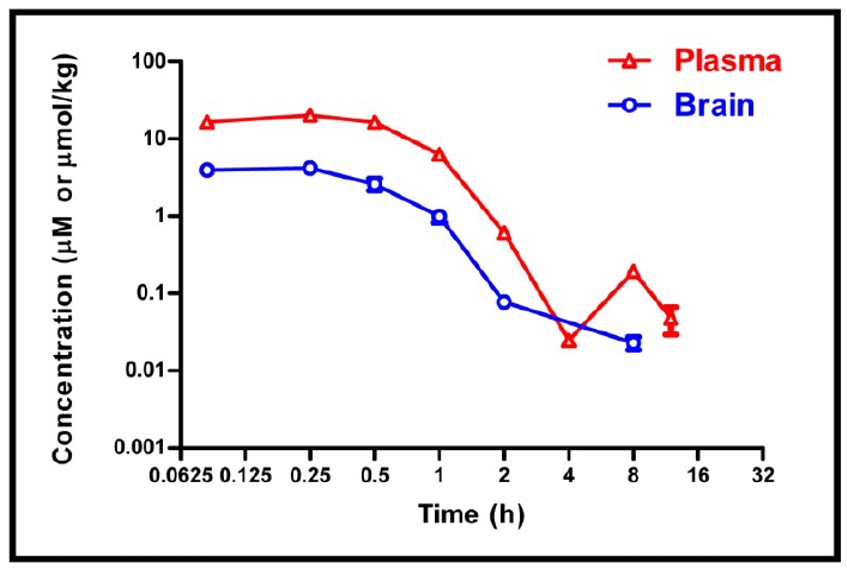
Figure 2. Mean plasma and brain concentration-time profiles of ML321 after an IP dose of 30 mg/kg in male C57BL/6 mice (N = 3).
In vitro and vivo activity - Prior art comparison
Summary /
A recent group of publications appeared on a series of N-(1-benzylpiperidin-4-yl)pyridazine 3-amines exemplified by the clinical candidate JNJ-37822681 showing D inhibition (Langlois 2012). However, JNJ-37822681 is not very selective between D2 (Ki = 158 nM) and D3 (Ki = 1,159 nM) in displacement assays. Another compound L741,626, a close analog of Haloperidol, had claimed selectivity of ~10–15 fold in D2 and D3 displacement assays (Grundt 2007). This compound showed a 3-fold selectivity in functional D2 and D3 β-arrestin assays. The binding selectivity was significantly improved through several rounds of medicinal chemistry efforts to obtain analogs 57 and 58 which was observed to have D2 vs. D3 selectivity greater than 100 fold (Vangveravong, 2010). However, no further reports related to ADME profiling and in vivo animal data was reported. It is well known that 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and its derivatives are potential neurotoxins and could be a reason for lack of in vivo data for this chemical series. Hence, the reference compounds 57 and 58 were resynthesized in house and assayed side-by-side with the probe ML321. A stability and GPCR panel inhibition profile for compounds 58 and ML321 were also assessed. Compound 58 was observed to have a 60-fold D2 vs. D3 selectivity in the β-arrestin functional assay and 48.6-fold D2 vs. D3 selectivity in the binding assay (Table 4). However, it showed a very short half-life time of 2.4 min in rat liver microsomal test. Results of the GPCR panel inhibition profile also showed that compound 58 at 10 μM is very promiscuous, targeting a large number of receptors with > 50% inhibition (Table 5). Not only did it inhibit dopamine receptors 2–4, but also targeted Sigma 1–2, SERT, MOR and Alpha2 A-B with > 90% inhibition. In contrast, ML321 at 10 μM gave a more selective profile; with > 90% inhibition only against the D2 receptor while the rest (5-HT2C, 5-HT7, D3) were only ~ 60% inhibition. Until this probe, it was very difficult to interpret data using existing compounds in ex vivo preparation because so many of them are “contaminated” by influence from the D3 receptor or the D4 receptor; both of which is antagonized by all existing molecules. Although ML321 (and members of its chemical series) is less potent than the compound 58, ML321 showed a good D2 vs. D3 receptor selectivity and good ADME and PK properties with fewer chemical scaffold liabilities. Therefore, probe ML321 provides sufficient selectivity towards D2 giving scientist a tool to better understand it contributions to dopamine signaling pathways both in vitro and in vivo (Xiao 2010).
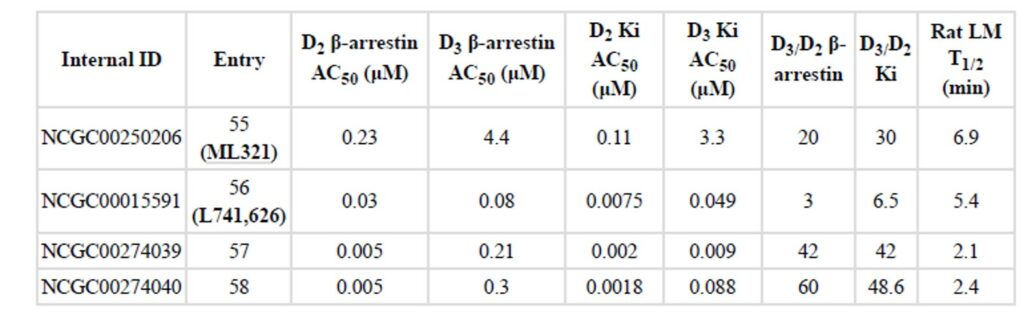
Table 4. Comparison of dopamine receptor inhibition assay for ML321 and current D antagonists.
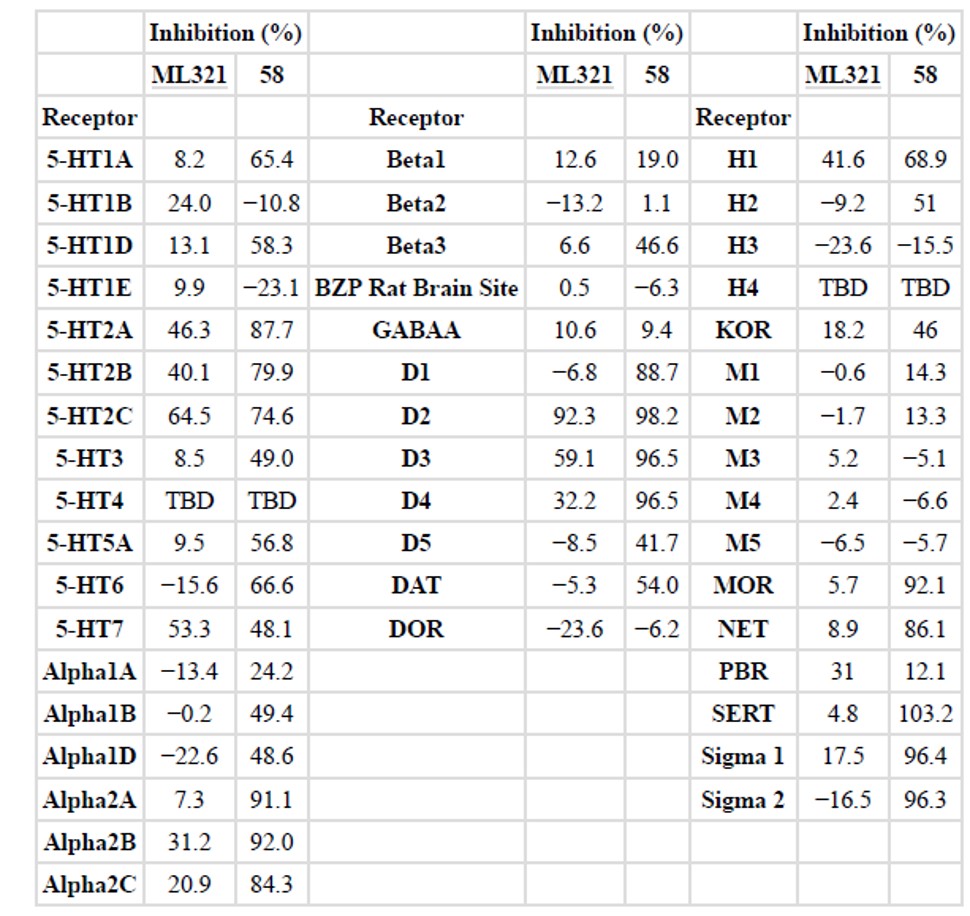
Table 5. GPCR panel screening for ML321 and compound 58. Data represent mean % inhibition (N = 4 determinations) for compound tested at receptor sub-types. > 50% of inhibition (marked red) is considered as significant inhibition. In cases where negative inhibition (−) is seen, this represents a stimulation of binding. Occasionally, compounds at high concentrations will non-specifically increase binding. The default concentration for primary binding experiments is 10 μM. Receptor binding profiles was generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2008-025C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA.
References
- HTS Assay for Allosteric Antagonists of the Human D2 Dopamine Receptor: Antagonists Summary
- Xiao J, Free RB, Barnaeva E, et al. Discovery, optimization, and characterization of a novel series of dopamine D2 versus D3 receptor selective antagonists. 2012 Dec 25 [Updated 2013 Sep 3]. In: Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010
- Kuhar MJ, Couceyro PR, Lambert P. Dopamine Receptors. In: Siegel G, Agranoff B, Albers R, et al., editors. Basic Neurochemistry: Molecular Cellular and Medical Aspects. 6th edition. Philadelphia: Lippincott-Raven; 1999. NCBI Bookshelf ID: NBK27980
- Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63(1):182-217. doi:10.1124/pr.110.002642
- Seeman P. Dopamine Receptors: Clinical Correlates. In: Sibley D, editor. Psychopharmacology – 4th Generation of Progress. New York: Nature Publishing Group; 2000
- Cho DI, Zheng M, Kim KM. Current Perspectives on the Selective Regulation of Dopamine D2 and D3 Receptors. Arch Pharm Res. 2010;33(10):1521–1538
- Miyake N, Miyamoto S, Jarskog LF. New seratonin/dopamine antagonists for the treatment of schizophrenia: are we making real progress? Clin Schizophr Relat Psychoses. 2012;6(3):122–133
- Ginovart N, Kapur S. Role of Dopamine D2 receptors for antipsychotic activity. Handb Exp Pharmacol. 2012;212:27–52
- Le Foll B, Gallo A, Le Strat Y, Gorwood P. Genetics of dopamine receptors and drug addiction: a comprehensive review. Behav Pharmacol. 2009;20(1):1–17
- Langlois X, Megens A, Lavreysen H, Atack J, Cik M, te Riele P, Peeters L, Wouters R, Vermeire J, Hendrickx H, Macdonald G, De Bruyn M. Pharmacology of JNJ-37822681, a specific and fast-dissociating D2 antagonist for the treatment of schizophrenia. J Pharmacol Exp Ther. 2012;342(1):91–105
- Grundt P, Husband SL, Luedtke RR, Taylor M, Newman AH. Analogues of the dopamine D2 receptor antagonist L741,626: Binding, function, and SAR. Bioorg Med Chem Lett. 2007;17(3):745–749
- Vangveravong S, Taylor M, Xu J, Cui J, Calvin W, Babic S, Luedtke RR, Mach RH. Synthesis and characterization of selective dopamine D2 receptor antagonists. 2. Azaindole, benzofuran, and benzothiophene analogs of L-741,626. Bioorg Med Chem. 2010;18(14):5291–5300